

Desde el descubrimiento del principio activo del medicamento para hipertensión hoy conocido como Valsartán [1] hasta la aprobación de la Food & Drug Administration (FDA) [2] transcurrieron 10 años. El caso de Valsartán es representativo de la gran mayoría de los medicamentos desarrollados hasta comienzos del siglo XXI, para los cuales tomaba alrededor de 10 a 15 años lograr su aprobación desde el descubrimiento del compuesto químico. Justamente la primera etapa, el descubrimiento de fármacos, es una de las que más consume tiempo en todo el proceso de desarrollo farmacéutico. Sin embargo, actualmente la investigación en farmacología tiene un aliado que ha contribuido de manera importante en el desarrollo de medicamentos y que ha tenido un rol destacado durante la pandemia: la química computacional.
El desarrollo de un medicamento comienza con la exploración de miles de compuestos químicos, que en farmacología tradicional usualmente provienen de plantas o microorganismos. Estos miles de compuestos químicos deben ser primeramente evaluados por medio de variadas técnicas de análisis como diferentes tipos de microscopía y cromatografía, difracción de rayos X, espectrometría, entre otras. Al mismo tiempo, los compuestos químicos debían ser puestos a prueba en ensayos de laboratorio en cultivos celulares y en organismos modelo de experimentación científica. Si bien esta metodología aún está en uso, la incorporación de métodos bioinformáticos y computación de alta capacidad de procesamiento ha permitido acelerar la etapa de descubrimiento de fármacos, ahorrando así tiempo, recursos monetarios e insumos químicos.
Diseño de fármacos in silico
El descubrimiento de fármacos asistido por la computación, denominado in silico, es un ciclo iterativo que comienza con grandes bibliotecas químicas en busca de la identificación de compuestos con potencial farmacológico prometedor. Para hacer más eficiente el descubrimiento de fármacos, en las últimas décadas se ha recurrido a computadores con alta capacidad de procesamiento bioinformático para analizar los compuestos químicos de las bibliotecas. El rol del investigador/a comienza con entregar al sistema la información teórica y empírica para definir los parámetros de la búsqueda y seleccionar a los candidatos más adecuados. Así, en cada iteración se acota la búsqueda y se genera una nueva biblioteca, con el fin de posteriormente validar los resultados mediante ensayos clínicos.
Estas técnicas computacionales (Figura 1) ayudan a analizar billones de datos de forma imparcial, a perfeccionar el diseño de nuevos compuestos y a conseguir una evaluación de un fármaco candidato para el diseño de medicamentos en menos tiempo. Los avances de la computación y la química computacional han propiciado la generación de sistemas que permiten calcular parámetros clave como la geometría y la energía molecular a partir de un gran volumen de datos, tal que sería imposible de procesar de otra manera. Los datos generados por estos sistemas tienen una amplia aplicación en la investigación experimental, de modo que la información que entregan es utilizada por investigadores tanto para la interpretación de los resultados obtenidos, la planificación de futuros experimentos, así como también para deducir información que no es asequible experimentalmente.

Caso de estudio: inhibidores de SARS-CoV-2
La pandemia iniciada en Wuhan (China) en 2019 ha causado un gran número de muertes y ha infectado a personas de todo el mundo debido, en parte, a la ausencia de una terapia eficaz contra el Coronavirus 2 del Síndrome Respiratorio Agudo Severo (SARS-CoV-2) [3]. En nuestro país, el primer caso se reportó el 3 de marzo de 2020, y casi una semana después la Organización Mundial de la Salud declaró oficialmente el estado de pandemia [4].
En consecuencia, múltiples equipos de investigación en países desarrollados y en vías de desarrollo se volcaron a la generación de vacunas o antivirales contra COVID-19, el síndrome respiratorio provocado por el virus SARS-CoV-2, para encontrar soluciones en el corto y mediano plazo.
Las estrategias farmacológicas actuales contra COVID-19 buscan prevenir las graves complicaciones causadas por la infección por SARS-CoV-2. A la fecha se han desarrollado terapias con anticuerpos monoclonales para uso de emergencia (etesevimab en Estados Unidos). No obstante, todavía no hay un tratamiento completamente seguro y efectivo, lo que dificulta el control de la pandemia. En este contexto, muchos gobiernos, instituciones médicas y grupos de investigación científica han explorado el historial de brotes infecciosos similares y han puesto a prueba estos tratamientos. Los resultados han sido prometedores, pero hasta ahora no concluyentes. Algunos de ellos son cloroquina, hidroxicloroquina, camostat, nafamostat, umifenovir, tenofovir, ramdesidir, sofosbuvir, galidesivir, lopinavir y indinavir, que han demostrado in silico e in vitro cierto grado de inhibición del SARS-CoV-2 [5–15].
Los fármacos mencionados están siendo utilizados para contrarrestar la actividad viral, pero aún no se tiene un entendimiento acabado de los efectos secundarios. Por lo tanto, es necesario continuar descubriendo nuevos antivirales contra SARS-CoV-2.
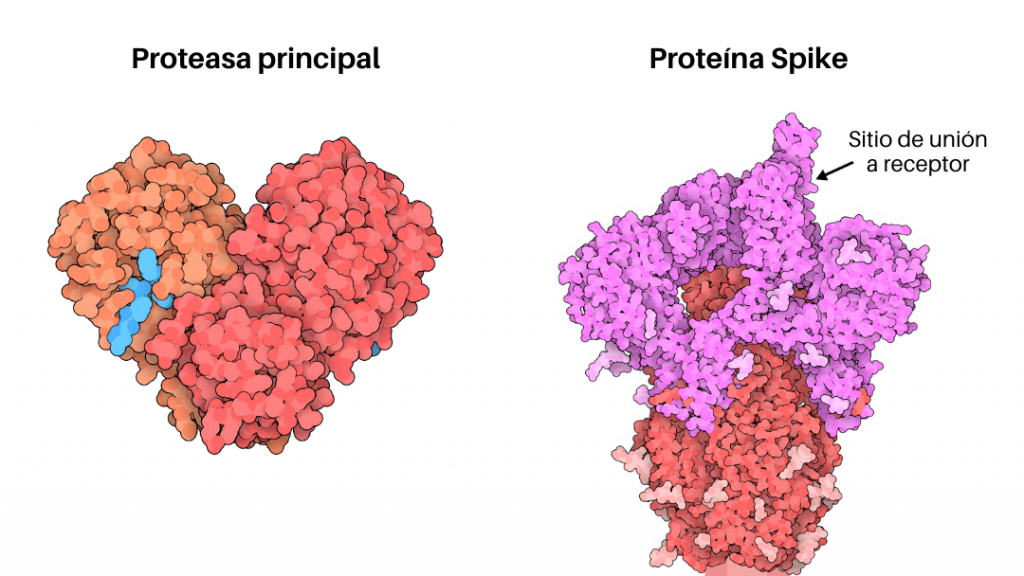
Con el fin de acelerar la búsqueda, el enfoque que se adopta es un diseño racional dirigido específicamente a las proteínas del SARS-CoV-2 para interrumpir su ciclo vital y detener su reproducción en el organismo a expensas de los recursos del hospedero. Existen aproximadamente 25 proteínas que el virus necesita para replicarse e infectar a los humanos y estas proteínas son, en otras palabras, posibles blancos terapéuticos. Dentro de estos blancos terapéuticos los más estudiados son la proteasa principal (Mpro), la glicoproteína Spike (Figura 2) y las proteínas TMPRSS2. Es imperante para la salud de todos nosotros encontrar fármacos que se unan a estas proteínas víricas e impidan su funcionamiento.
En la urgente tarea de descubrimiento de nuevos fármacos para SARS-CoV-2, los supercomputadores y la química computacional han jugado un rol muy importante para el descubrimiento de nuevos inhibidores en los diferentes blancos terapéuticos nombrados. Para alcanzar este objetivo, los autores Phimister EG et al 2020 [17] en su artículo How to Discover Antiviral Drugs Quickly, proponen un protocolo que consiste en imitar a la naturaleza para el descubrimiento de fármacos. Este protocolo se basa en un análisis de la estructura molecular utilizando la metodología de computación paralela y distribuida (Figura 3). El descubrimiento de fármacos basado en la estructura ha sido importante para encontrar fármacos antivirales; un ejemplo es el nelfinavir descubierto en la década de 1990 para tratar la infección por el virus de la inmunodeficiencia humana (VIH) [18].
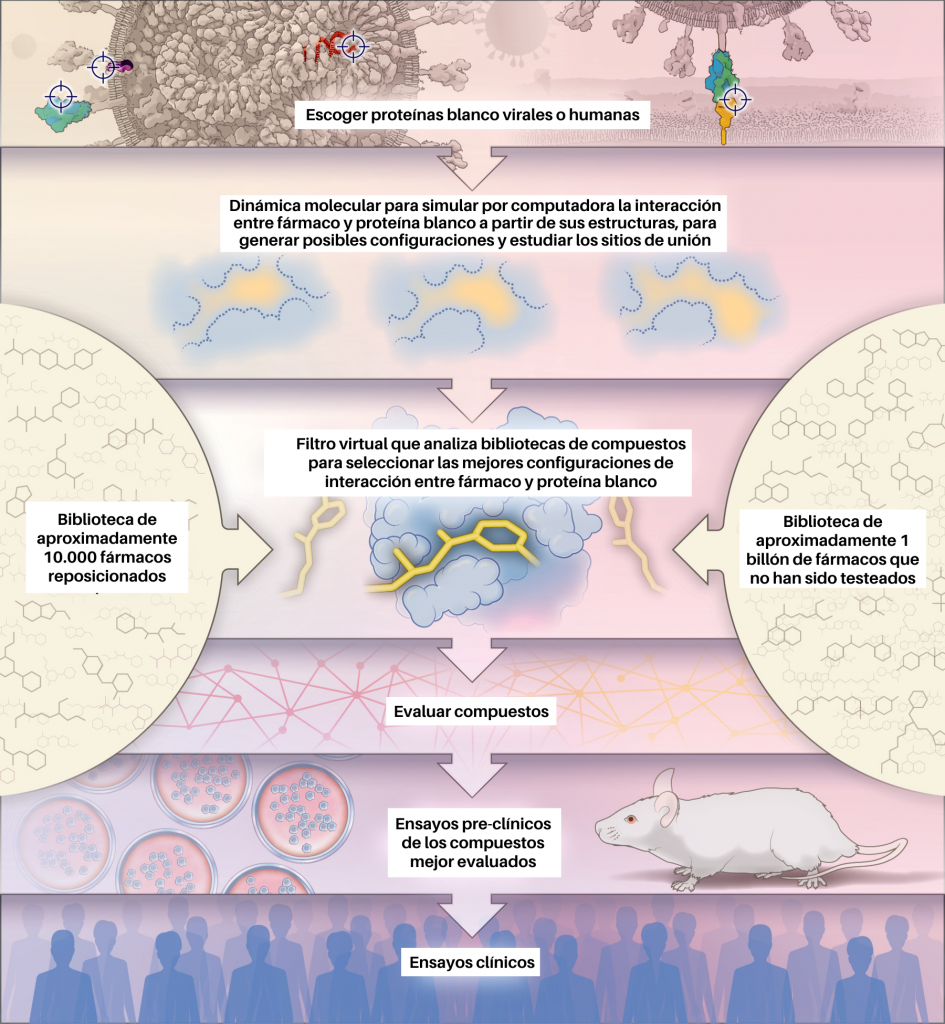
En el descubrimiento de fármacos basado en estructura los computadores «acoplan» los compuestos de prueba en sitios de unión en modelos tridimensionales de las proteínas objetivo. La afinidad de unión de los compuestos con las proteínas objetivo se calcula mediante ecuaciones físicas que cuantifican las interacciones entre el fármaco y su blanco terapéutico. Luego, los compuestos candidatos se ordenan según un puntaje que incluye la afinidad de unión y otros parámetros farmacocinéticos.
A modo de ilustración, en Yáñez O. et al 2021 [19] se utilizó como modelo computacional la estructura cristalizada de la enzima Mpro de SARS-CoV-2 en búsqueda de posibles inhibidores. Los compuestos evaluados fueron 26 ligandos sintéticos derivados de cumarinas y quinolinas, un tipo de compuestos que ha sido utilizado contra SARS-CoV-2 [15] y otros virus [19, 20]. El objetivo de la evaluación fue encontrar los compuestos más apropiados para inhibir la actividad del coronavirus, por lo cual se analizaron por acoplamiento molecular rígido, cálculo de eficiencia, predicción de propiedades farmacológicas y toxicológicas, y simulación por dinámica molecular, realizando así una predicción de la energía libre de unión. A partir del análisis, el equipo de investigación identificó 6 compuestos (Figura 4) con alta afinidad de unión por Mpro y baja toxicidad.

El siguiente paso en el descubrimiento de fármacos asistido por computadoras es someter a los compuestos con mejores puntajes a pruebas experimentales en células y modelos animales para comprobar si realmente se unen al blanco terapéutico y tienen los efectos posteriores requeridos, como en el caso de SARS-CoV-2 es detener la infectividad viral.
El diseño in silico de fármacos está en continua evolución, aplicando una gran variedad de métodos computacionales para identificar las complejas relaciones existentes entre estructuras moleculares y actividades biológicas, en términos de interacciones entre los átomos constituyentes. La introducción de nuevos métodos teóricos y de computadores de alto rendimiento o cuánticos, ayudarán sin duda a la obtención de nuevos fármacos cada vez más potentes, selectivos y específicos con sus blancos terapéuticos.
Artículo colaborativo:
Dr. Osvaldo Yáñez Osses, investigador postdoctoral CENDHY
Lorena Díaz Hemard, coordinadora científica y de comunicaciones CENDHY
Dr. Javier O. Morales, Director de CENDHY
Dr. Eduardo Soto-Bustamante, Subdirector de CENDHY
Referencias
[1] John V. Duncia, David J. Carini, Andrew T. Chiu, Alexander L. Johnson, William A. Price, Pancras C. Wong, Ruth R. Wexler, Pieter B.M.W.M. Timmermans. The Discovery of DuP 753, a Potent, Orally Active Nonpeptide Angiotensin I1 Receptor Antagonist. Medical Research Reviews 1992 Vol. 12, No. 2, 149-191. DOI: https://doi.org/10.1002/med.2610120203
[2] Drug Approval Package. Diovan (Valsartan) Capsules & Tablets. Food & Drug Administration (FDA).
[3] COVID-19 Map – Johns Hopkins Coronavirus Resource Center Available online: https://coronavirus.jhu.edu/map.html
[4] Cucinotta D, Vanelli M. WHO Declares COVID-19 a Pandemic. Acta Biomed 2020 91(1): 157-160. DOI: 10.23750/abm.v91i1.9397
[5] Elfiky AA. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. Disponible para descarga gratuita.
[6] Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N.-H, Nitsche A, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271-280.e8. Disponible para descarga gratuita.
[7] Hirota M, Shimosegawa T, Kitamura K, Takeda K, Takeyama Y, Mayumi T, Ito T, Takenaka M, Iwasaki E, Sawano H, et al. Continuous regional arterial infusion versus intravenous administration of the protease inhibitor nafamostat mesilate for predicted severe acute pancreatitis: a multicenter, randomized, open-label, phase 2 trial. J. Gastroenterol. 2020, 55, 342–352. Disponible para descarga gratuita.
[8] Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J, Matsuda Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532 LP – 6539, Disponible para descarga gratuita.
[9] Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Crit. Care 2020. Disponible para descarga gratuita.
[10] Mulangu S, Dodd LE, Davey RT, Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. Disponible para descarga gratuita.
[11] Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, Feldt T, Green G, Green ML, Lescure F.-X, et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. Disponible para descarga gratuita.
[12] Chu CM, Cheng VCC, Hung IFN, Wong MML, Chan KH, Chan KS, Kao RYT, Poon LLM, Wong CLP, Guan Y, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax 2004, 59, 252 LP – 256. Disponible para descarga gratuita.
[13] Deng L, Li C, Zeng Q, Liu X, Li X, Zhang H, Hong Z, Xia J. Arbidol combined with LPV/r versus LPV/r alone against Coronavirus Disease 2019: A retrospective cohort study. J. Infect. 2020. Disponible para descarga gratuita.
[14] Sissoko D, Laouenan C, Folkesson E, M’Lebing AB, Beavogui AH, Baize S, Camara AM, Maes P, Shepherd S, Danel C, et al. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016, 13, 1–36. Disponible para descarga gratuita.
[15] McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. Disponible para descarga gratuita.
[16] Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 3405, eabb3405. Disponible para descarga gratuita.
[17] Phimister, E.G.; Parks, J.M.; Smith, J.C. How to Discover Antiviral Drugs Quickly. N Engl J Med 2020; 382:2261-2264. Disponible para descarga gratuita.
[18] Patick AK & Potts KE. Protease inhibitors as antiviral agents. Clinical Microbiology Reviews 1998, 11(4), 614–627. Disponible para descarga gratuita.
[19] Yañez O, Osorio MI, Uriarte E, Areche C, Tiznado W, Pérez-Donoso JM, García-Beltrán O, González-Nilo F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Frontiers in Chemistry. 2021, 8:1273. Disponible en Publicaciones.
[20] Mishra S, Pandey A, Manvati S. Coumarin: An emerging antiviral agent. Heliyon. 2020 6(1):e03217. Disponible para descarga gratuita.
0 comentarios